Our retinal projects are organised around five major themes:
Funded by the Macular Society, we have been able to create a disease model in the lab which we can use for various studies
Age related macular degeneration (AMD) is the commonest cause of blindness in the developed world. There are two manifestations of advanced AMD resulting in loss of vision: dry and wet; the dry type being the most common and with no treatment available. Understanding what happens in AMD affected retina is a crucial basis for developing therapies. Funded by the Macular Society, our lab generated a cell model of AMD by turning skin cells from people affected by AMD into retinal pigment epithelium (RPE) cells, the principle cell type that gets damaged and lost during the course of AMD (Hallam et al. 2017, Stem Cells. 2017 Nov;35(11):2305-2320). Our work has shown that AMD-RPE cells have expanded and less functional lysosomal compartments, which are organelles that are involved in digestion of unwanted intracellular materials (autophagy) (Cerniauskas et al. 2020, Stem Cells Transl Med. 2020 Dec;9(12):1585-1603). We found that lysosomal membranes are fragile and this may cause the leakage of their crucial digestive enzymes and their deposition into drusen. Drusen are build-ups of waste products under the RPE and large accumulations of drusen are the defining feature of macular degeneration. We have also shown that the probable cause of the lysosomal deficiency is uncontrolled activation of the complement system, which is part of the immune system that clears pathogens. Treatment with a specific complement inhibitor that stops this activity helped to stop the progression of AMD features in AMD-RPE cells, which are promising results suggesting new treatment options for AMD
Our further work, funded by the Macular Society, has shown that AMD-RPE cells produce stress markers that are moved outside the cells in tiny vesicles called exosomes, which help cells to communicate with each other (Kurzawa-Akanbi at al. in preparation). We are able to isolate these vesicles and analyse their contents. We found that AMD-RPE produce around twice as many exosomes compared to RPE cells derived from people without AMD. AMD-RPE exosomes are very harmful to healthy RPE cells and make them develop AMD-like features, which makes us believe they are involved in the progression of AMD. We are now in a process of careful examination of AMD-RPE exosomes contents, which will allow us to pinpoint specific exosomal markers that could potentially be used to detect AMD. As exosomes are tiny, they can enter the bloodstream and we are hoping to be able to detect those AMD-RPE stress related exosomal markers in the patients’ blood, which could give us means to spot AMD early and predict the progression of the disease (dry or wet type).
As our studies implicate faulty autophagy in macular degeneration, we are looking into other macular diseases that may be due to similar cellular defects. Mutations in DRAM2, which is an autophagy regulator, are known to cause retinal degeneration with early macular cone photoreceptor involvement. Our lab generated induced pluripotent stem cell lines (iPSC) from patient skin cells with DRAM2 mutations. We have been able to erase these mutations using the CRISPR technology (CRISPR) and produce controls for the disease mutation-bearing cell lines (Rozaliya Tsikandelova ongoing work). Using these stem cells, we are generating retinal organoids (“retinas in a dish”) models and RPE monolayers that will allow us to study the role of mutant DRAM2 in autophagy dysfunction in the RPE cells and the photoreceptors.
Transforming ophthalmic research
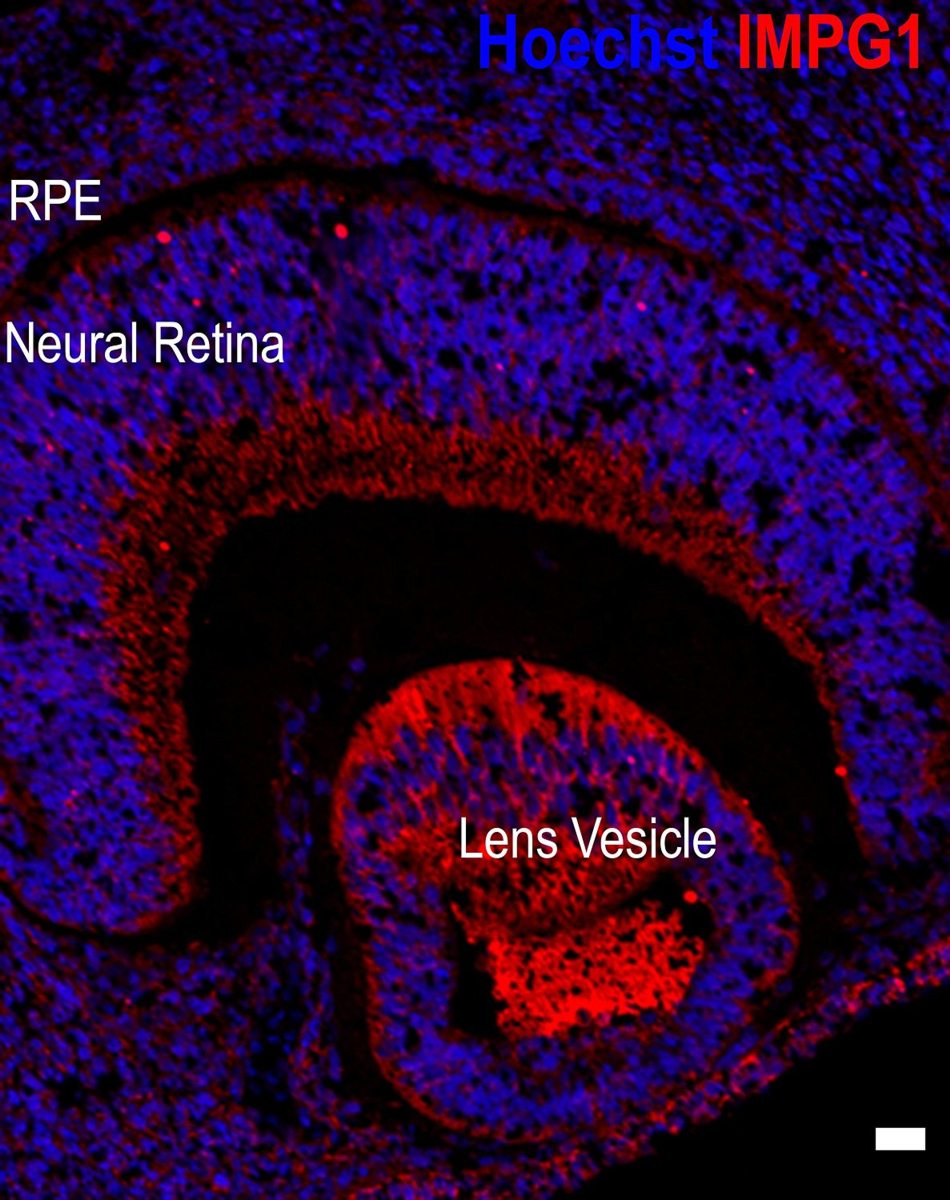
The generation of human retinal tissue from stem cells under laboratory conditions has been a pioneering breakthrough which has transformed the field of ophthalmic research. In the last five years, our group has developed robust methods for the large-scale generation of retinal organoids (retina in a dish) from human embryonic and human induced pluripotent stem cells. These mini retinas in a dish contain all retinal cell types and display the same five layered structure as known from adult retina. Moreover, they exhibit the most crucial function of the retina, light sensitivity. Our group is using these mini retinas for:
Retinal organoids derived from human pluripotent stem cells.
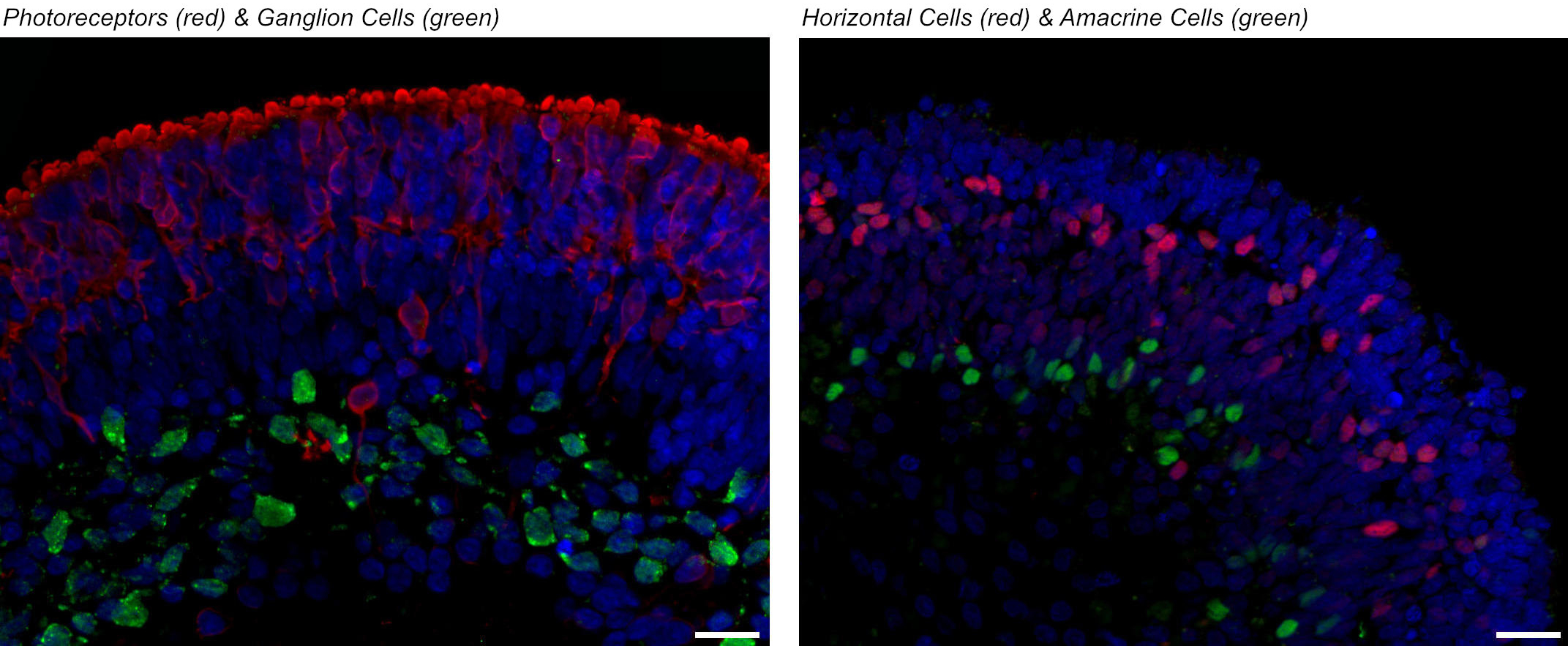
These projects have benefited from multiple funding awards (European Research Council, Retina UK and NCR3Rs) and close collaborations with Prof. Evelyne Sernagor at the Bioscience Institute (Newcastle University) and Dr Gerrit Hilgen (Northumbria University).
Our approach to deriving patient specific induced pluripotent stem cells (iPSC)
Retinitis pigmentosa (RP) is the most common inherited retinal disease characterized by progressive degeneration of photoreceptors leading to night vision loss, restricted visual field until eventual blindness. The prevalence of RP is around 1 in 4000 and there are over 1.5 million RP patients worldwide. Mutations in the pre-mRNA processing factor (PRPF) genes encoding the core spliceosome components account for 15-20% of autosomal dominant RP cases. Using reprogramming approaches and CRISPR/Cas9 technologies our group has generated patient specific iPSC lines from patients with PRPF31 and PRPF8 mutations. Our work has shown severe RPE defects resulting from PRPF31 mutations, including global spliceosome dysregulation, disrupted apical-basal polarity, reduced trans-epithelial resistance, compromised phagocytic capacity, decreased cilia length and incidence. Disrupted cilia morphology also occurred in patient-derived photoreceptors, associated with progressive degeneration and cellular stress (Buskin et al. Nat Commun. 2018 Oct 12;9(1):4234).
In collaboration with Prof. Robin Ali and his team, we are assessing the potential of gene therapy to restore the molecular and cellular dysfunction observed in RPRPF31 retinal cells. Funded by the MRC and in collaboration with research groups led by Prof. Colin Johnson (University of Leeds), Prof. Robin Ali (King’s College, London) Dr. Sina Mozaffari-Jovin (Max Planck Institute, Gottingen) and Dr Sushma Nagaraja-Grellscheid (University of Bergen), we are investigating the impact of PRPF8 mutations on RPE cells and whether these could be restored by a combination of gene therapy and small molecule based therapies.
The potential to investigate pathological changes in the human eye
Retinoblastoma is a childhood cancer of the developing retina, the light-detecting tissue of the eye. Biallelic mutations in RB1 gene encoding pRB (retinoblastoma protein) accounts for ~98 % of retinoblastoma cases in young children (up to five years old).
The palette of pRB degenerative mutations that define malignant signature and the demand to establish enhanced clinical protocols of treatment that not only ensures child’s survival but also normal development post-treatment hence low toxicity with high efficacy, cannot make a more compelling case for further studies.
Well established in our laboratory methodology for retinal organoids formation and enormous potential of this 3D model to investigate pathological changes in the human eye encouraged us to establish in vitro model of retinoblastoma, initiated by biallelic inactivation of RB1. This project benefited from Fight from Sight funding and close collaborations with Consultant Ophthalmologist, Mr. Manoj Parulekar.
To obtain the model that resembles patient-specific genomic background in combination with a hereditary mutation in the pRB gene, we generated iPSC lines from two patients heterozygous for the mutations; c.2082delC or c.607+1G>C. We employed CRISPR/Cas9 gene-editing technique to introduce mutation in the wild-type allele of RB1 gene or correct the mutation. Retinal organoids made from patient iPSCs lacking expression of RB1 gene displayed the hallmarks of retinoblastoma tumours and responded in a predictable manner to key chemotherapeutics substances used for treatment of the disease. Funded by the Little Princess Trust, we are undertaking screening of 39 compounds, with promising leads to take forward in pharmacogenetic studies.
There is a need for research into replacement of photoreceptors
Our retina does not show significant regenerative capacity; hence, there is a need for research into replacement of photoreceptors in patients with retinal disease. Our aim is to study and improve photoreceptors transplantation with the aim of developing therapeutic interventions to restore vision at advanced stages of retinal degeneration.
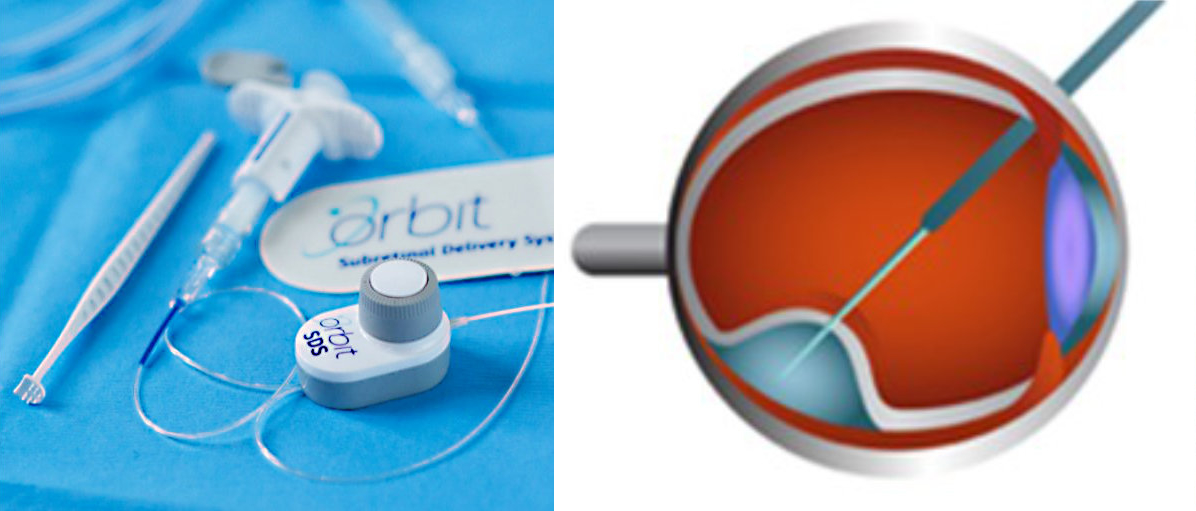
With the advances made in differentiation of human pluripotent stem cells (hPSCs), we are able to generate retinal organoids containing photoreceptors developing alongside neurons and Müller glial cells in a laminated structure resembling the native retina. We have developed pre-clinical tools for enrichment of photoreceptors (Collin et al., 2016; Stem Cells. 2016 Feb;34(2):311-21), which we deliver as cell suspensions into the subretinal space (Collin et al. 2019; Stem Cells. 2019 May;37(5):609-622). We then perform behavioral and functional assays for assessing photoreceptor engraftment and vision restoration in animal models of retinal degeneration in close collaboration with Prof. Evelyne Sernagor (Newcastle University) and Dr. Gerrit Hilgen (Northumbria University). Funded by the MRC we are assessing the most optimal rod-cone transplant in animal models of retinal degeneration for treatment of advanced Retinitis Pigmentosa.
We are actively participating in current clinical trials
Our group is also actively participating in clinical trials of advanced therapies and hope to develop our own studies in due course. Professor Steel participated as a Phase 1 surgeon in the ground-breaking Ocata (now Astellas Institute of Regenerative Medicine) therapeutics trial of embryonic stem cell-derived RPE cell transplantation into the subretinal space in patients with Stargardt's macular dystrophy. He was the second UK surgeon to deliver subretinal gene therapy for dry AMD as part of the phase 1 FOCUS study with Gyroscope Therapeutics, and is now recruiting for their phase 2/3 EXPLORE and HORIZON studies. (https://www.gyroscopetx.com/clinical-trials/)
He is also one of only a few global surgeons trained in a novel suprachoroidal subretinal delivery technique, soon to be used in the FOCUS study, and anticipated to offer several advantages to conventional trans-vitreal delivery.
Professor Steel is developing links with other companies to bring these innovative new treatments to the clinic in the fastest possible time frame. He hopes soon also to participate in the AGTC gene therapy trial for X linked RP. ( http://ir.agtc.com/news-releases/news-release-details/agtc-announces-updated-development-plan-its-x-linked-retinitis)
Our aim is to understand how and when parts of our eyes form, function and interact
Vision loss can occur through the effect of faulty genes we inherit from our parents as well as the accumulation of damage and the effect of various diseases throughout our lives.
Our ability to prevent and treat vision loss is closely linked with our knowledge of how eyes form, how they work and when and what is likely to go wrong. The availability of tissue to study during pregnancy and through adulthood is very limited. Our group is in a unique position, having access to eyes through resources such as the HDBR, a collaboration between our University and University College London, which collects samples from aborted embryos and foetuses with the mother's consent, and NHSBT.
Our aim is to use these samples to understand how and when parts of our eyes form, function and interact, and look at the role of genes that cause loss of vision when faulty. This work commenced in 2011 and we have compiled images of and information on genes involved in the developing and adult eye, which we have reported in Mellough et al Development. 2019 Jan 29;146(2): dev169474). Funded by BBSRC, we have completed this work at the single cell level, generating new insights into the location of retinal progenitor cells during early human development.
Retinal Stem Cell Research
Biosciences Institute
Newcastle University, International Centre for Life, Central Parkway, Newcastle upon Tyne, NE1 3 BZ. United Kingdom
Tel: +44 (0)191 241 8688
Email:
majlinda.lako@ncl.ac.uk